


Statin Truth!
Anti-inflammatory Effects of Statin Drugs
Lewis Clarke, M.D., Ph.D.
All patients with brain injury and stroke are given statin drugs. However, science supporting therapeutic benefit is not convincing. Statin drugs lower cholesterol. Statin drugs also reduce formation of atherosclerotic plaque formation. To the general public and unfortunately to most physicians that means that vascular disease is the result of too much cholesterol floating around in the blood. They conceptualize this as if there is too much sugar in a glass of tea and it settles at the bottom, eventually plugging up the vessel. If this were the case, eliminating cholesterol should prevent heart disease, but it does not. Fifty percent of heart attacks and strokes occur in patients with normal cholesterol levels.
The cholesterol hypothesis of heart disease has long been thought to be untenable by biochemists. Rather, an unrestrained inflammatory cascade appears to be the etiology of the vascular lesions. The presence of cholesterol in these lesions is, as previously discussed, the body’s attempt at repair using cholesterol as a sort of plaster to restore a laminar, turbulence-free flow in a high pressurized pipe. Cholesterol is the repair molecule for all cells, not only in the brain and peripheral nervous system, but also in the cardiovascular system.
The cardiovascular benefits of statin drugs, therefore are not the result of lowered cholesterol, but rather the suppression of inflammation. Numerous recent papers have finally found their way into the general clinical literature demonstrating specific inflammation markers, high sensitivity C-reactive protein and homocysteine, are much more reliable barometers of atherosclerotic heart disease risk (Ridker, et. al., Lancet Aug 2017) (de Lemos, et. al. Circulation. 2017;135:2119-2132). Statin drugs reduce hs-CRP. In addition, statin drugs lower the levels of inflammatory cytokines, IL-1B, IL-6, IL-8, lipopolysaccharide induced cytokine activation, NFKappaB activation, MMP-9, COX-2, and tumor necrosis factor alpha, (Ridker, et. al., Lancet Aug 2017; McFarland, et. al., Mediators of Inflammation, Vol 2017 (2017), Article ID 2582745; Iwata, et.al., Clin Exp Immunol. 2012 May; 168(2): 234–240), and this has been demonstrated to occur in the CNS with inhibition of microglia phagocytic activity (Churchward and Todd, Mol Brain. 2014; 7: 85) by an action independent of its effect on cholesterol.
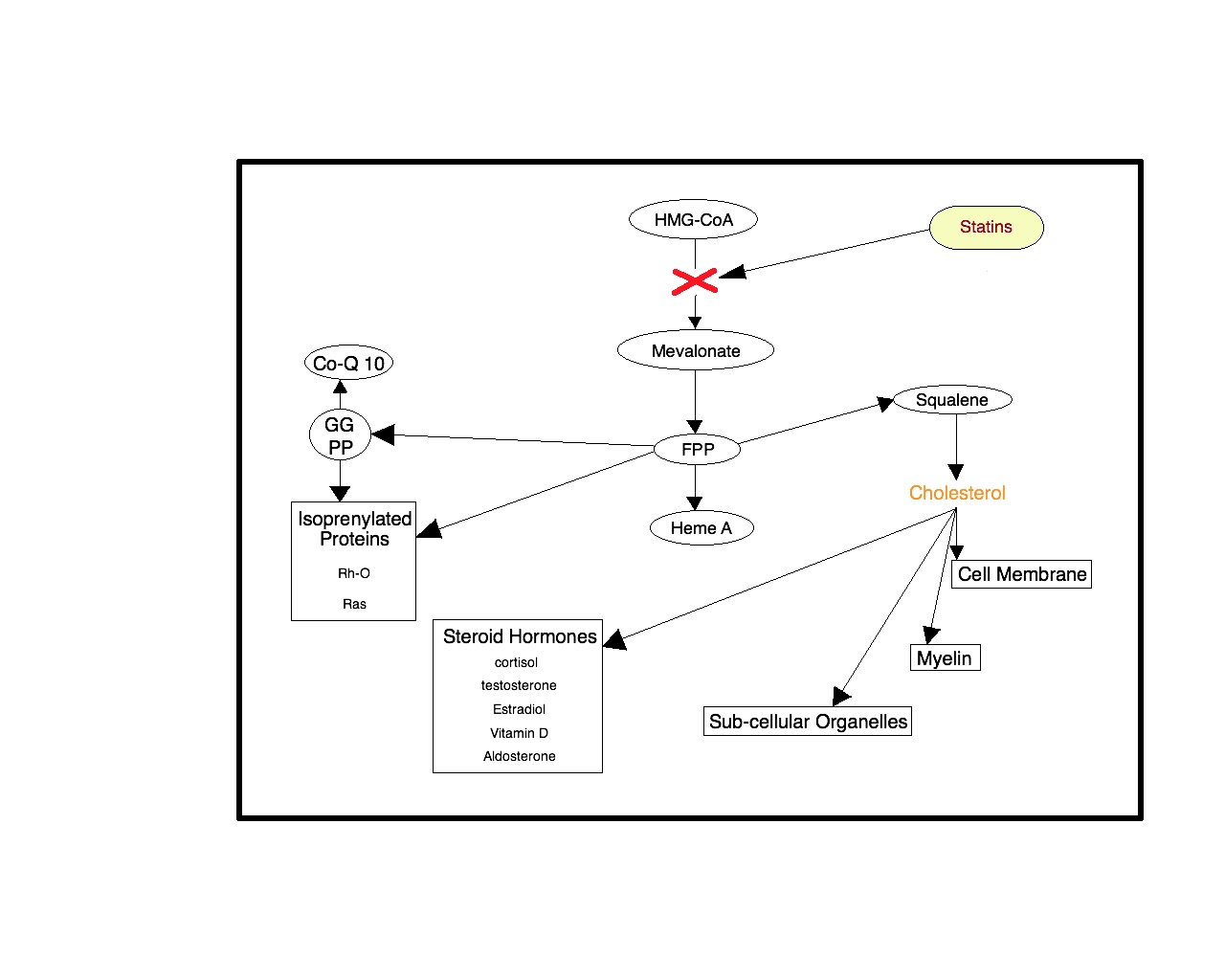
Using statins to suppress inflammation, however, is a double-edged sword. Statins inhibit cytokine production through inhibition of the mevalonate or isoprenoid pathway. So it appears that the only way to benefit from the anti-inflammatory effects of statin drugs is for the statin drug to inactivate this critical pathway. This is of paramount importance because when this pathway is inhibited by statins, there is a suppression of production of mitochondrial CoQ10 (thereby rendering mitochondria ineffectual in reining in the destructive sequence). There is also suppression of Rh-O and Ras proteins, suppression of myelin production and membrane and subcellular organelles, suppression of brain derived neurotrophic factor, nerve growth factors, neuron and oligodendrocyte formation from stem cells, suppression of mesenchymal stem cell production and glial progenitor cells, suppression and dysregulation of the neurosteroid production and sigma 1 receptor function, and alteration in dopamine and serotonin which indirectly alters sigma 1 function. This represents only an abbreviated list of the deleterious actions on recovery from neurologic injury effected by statin therapies.
In the figure below, the red ‘x’ is the location of action of statin drugs on the HMG-CoA reductase enzyme pathway whereby these drugs suppress the production of cholesterol. The mevalonate and isoprenoid pathways are critical for regenerative cellular functions and both are inhibited by the statin ‘x’. As discussed previously, all cell membranes derive from cholesterol, all the neurosteroids derive from cholesterol and these compounds stimulate the conversion of stem cells to all types of tissue cells including myelin, the insulation wrap of the nerve axon. The Rh-O protein unwinds the DNA-RNA structure and is essential to the cell’s reproduction of proteins and indeed essential to the reproduction of altogether new cells in its regenerative function. The Ras proteins switch on a variety of genes that begin cell replication, cell growth and differentiation. Therefore, the use of statin drugs following destructive cell processes like a heart attack or stroke would inhibit and suppress the very repair sequences programmed into every cell.
Original figure published in Dr. Clarke’s Chapter Preparing the Soil: Practical Cellular
Biochemistry for Regenerative Medicine in J. F. S. D. Lana et al. (eds.), Platelet-Rich Plasma,
Lecture Notes in Bioengineering, DOI: 10.1007/978-3-642-40117-6_3,
Springer-Verlag Berlin Heidelberg 2014
{kind=link}
Several conclusions can be made.
First, while statins may provide some neuroprotection in acute neurologic injury via anti-inflammatory effects, this benefit can only occur by statin inhibition of the mevalonate/isoprenoid cascade which then impedes mitochondrial energy production and neurorepair.
And as previously discussed, it is the mitochondria that arrests the progression of neurologic destruction.
Second, the suppression of neuron and glia and oligodendrocyte formation from progenitor stem cells and, in fact, the suppression of stem cell production itself makes the use of statins in the acute neurologic injury contraindicated since the process of neurogenesis is impaired.
Third, statin dysregulation of neurosteroid production and cholesterol binding on the sigma 1 receptors and the neurotransmitter alterations by statin drug therapy in the acute and the chronic degenerative neurologic diseases adversely affects neurogenesis and repair.
This is evidenced by the implication of the statin drugs in dementia and is not surprising since it is known that the neurosteroid DHEA converts stem cells to neurons in the hippocampus, the area of short term memory protein generation.
Finally, it is clear that there are endogenous mechanisms in the brain that are designed to protect and repair. When this neurochemistry is adequately supplemented following neurologic injury such as stroke, traumatic brain injury, concussion, or in chronic neurodegenerative diseases such as multiple sclerosis, Parkinson’s, Huntington’s, Alzheimer’s, and ALS, there can be neuroprotection and neurogenesis.
Summary
Once the brain is injured, whether by an ischemic or hemorrhagic stroke, or by sudden or by cumulative trauma, or by a gradual degenerative process as in Alzheimer’s, or by diseases as in M.S., Lou Gherig’s, and Parkinson’s, the mechanisms and nature of the destructive vicious circle are the same. However, there are three very important conclusions that are demonstrated in this discussion:
1). There are points at which neuropharmacologic interventions can break the cycles and mitigate further damage to the structure of the brain and prevent devastating loss of functions such as paralysis and cognitive decline.
2). As the injury sets into motion multiple closely intertwined progressively destructive processes each of which requiring individualized interventions, there can be no single drug that will effectively arrest the progression once the injury occurs. A heterogenous admixture of biochemical compounds must be designed to stop each of these destructive sequences separately and concurrently.